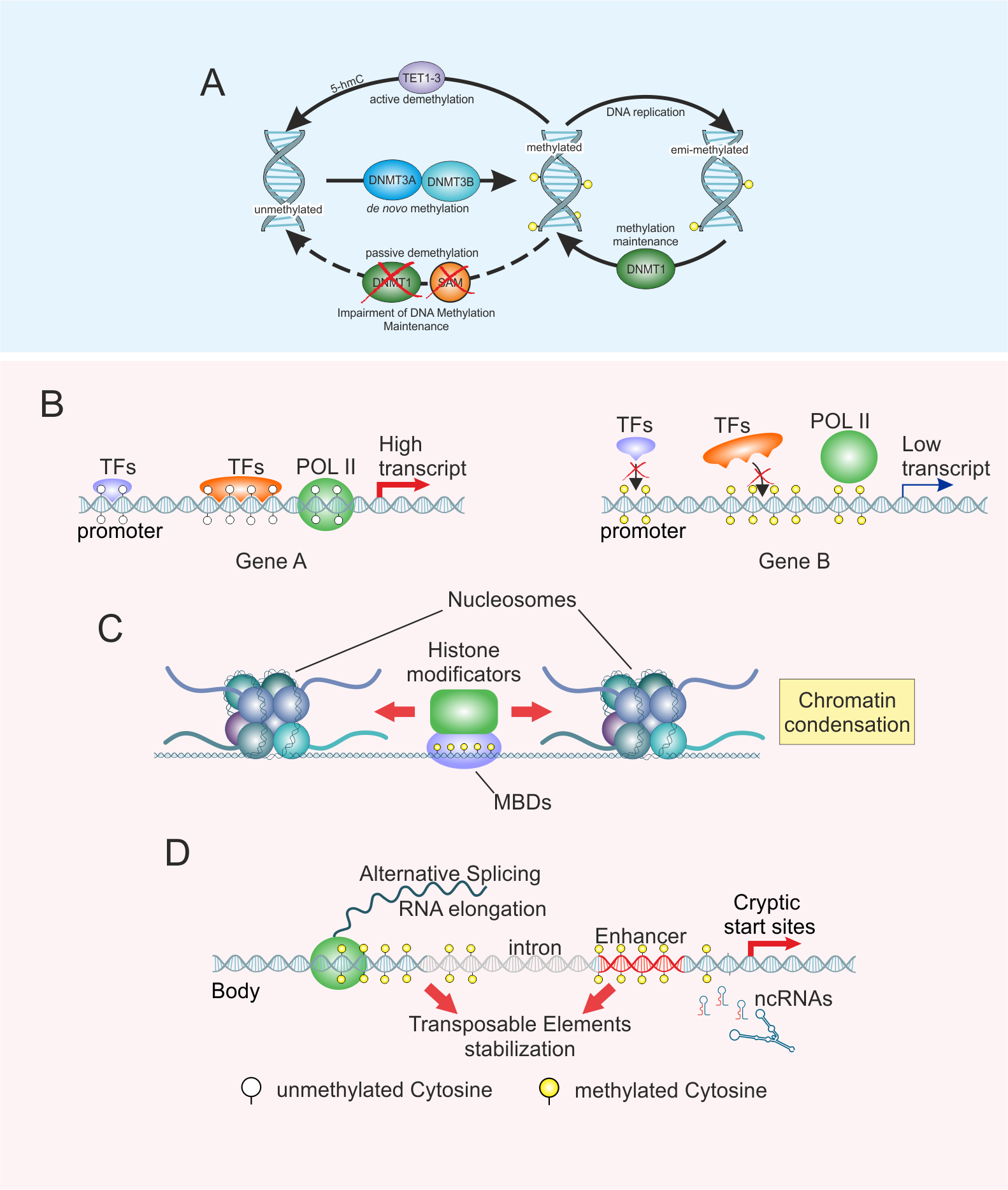
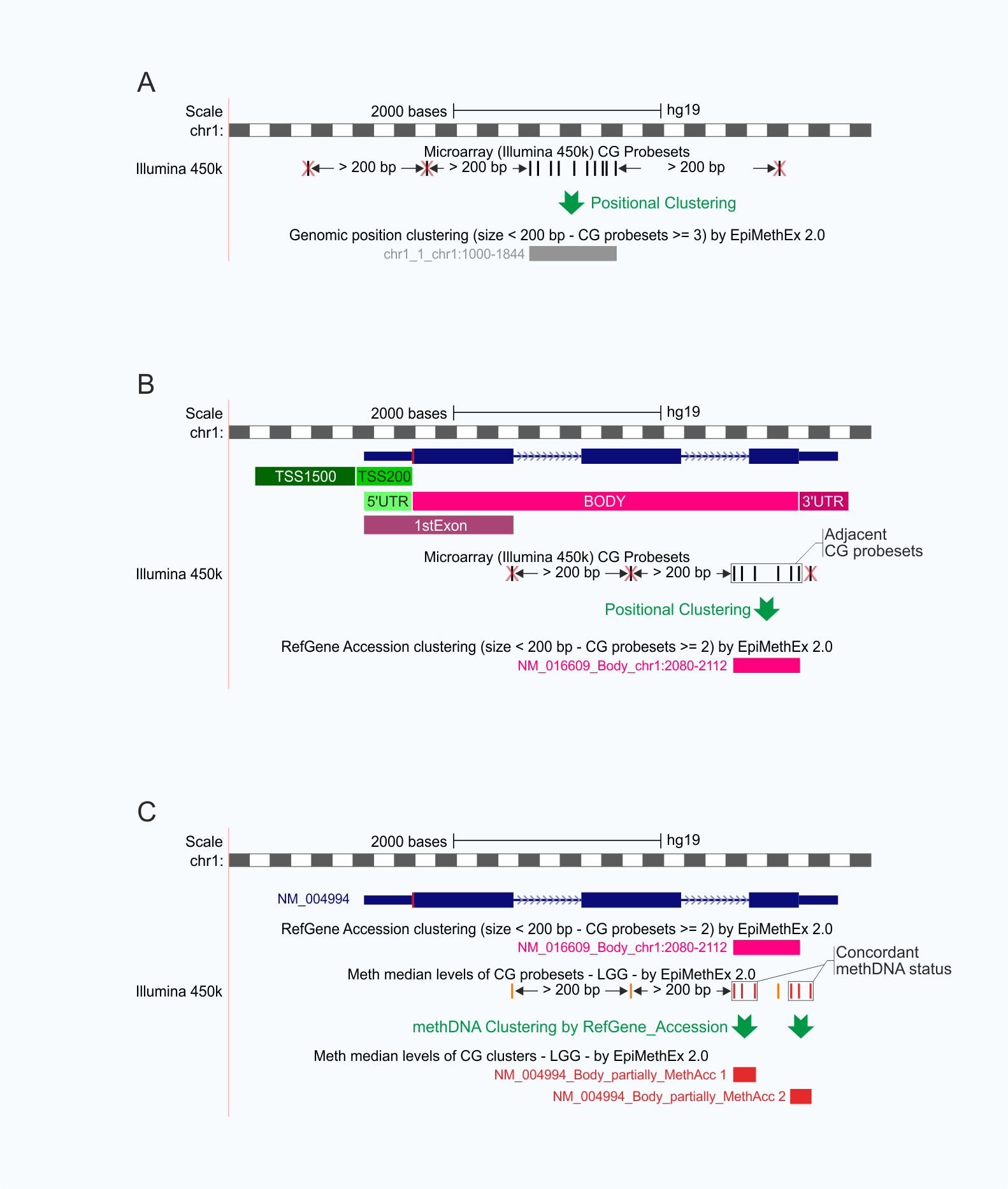
EpiMethEx 2.0 (Epigenetics Methylation Expression 2.0) is an R-based bioinformatic pipeline developed for the identification of both intragenic and intergenic DMRs using large-scale methDNA datasets.
The EpiMethEx 2.0, an improved version of our previous R-package EpiMethEx, is designed to perform clustering analysis of adjacent CG probesets (distance ≤ 200 bp, by default) based on their genomicPosition, RefGene_Name, and RefGene_Accession Annotations, as retrieved from Illumina HumanMethylation450 v1.2 Manifest (Figure 2A and 2B). The resulting positional CG clusters are subsequently used for additional clustering procedures according to concordant values of methDNA, Beta difference (Betadiff) between the comparison groups, and Correlation (Corr) values between methDNA status of each CG probeset and the expression levels of associated genes as follows:
methDNA CG clusters (Hypermethylated CG clusters: median methylation values > 0.6; Partially methylated CG clusters: 0.2 ≤ median methylation values ≤ 0.6; Hypomethylated CG clusters: median methylation values < 0.2);
Betadiff CG clusters (Weakly methylated CG clusters: 0.1 ≤ median beta value < 0.5; Strongly methylated CG clusters: median beta value ≥ 0.5; Weakly demethylated CG clusters: -0.1 ≤ median beta value > -0.5; Strongly demethylated CG clusters: median beta value ≤ -0.5);
Corr CG clusters (Weakly positively correlated CG clusters: 0 < r > 0.3; Moderately positively correlated CG clusters: 0.3 ≤ r ≥ 0.7; Strongly positively correlated CG clusters: r > 0.7; Weakly negatively correlated CG clusters: -0.3 ≤ r < 0; Moderately negatively correlated CG clusters: -0.7 ≤ r < -0.3; Strongly negatively correlated CG clusters: r < -0.7).
EpiMethEx 2.0 scripts also enable a comprehensive analysis by clustering the CG probesets of identified DMRs, here indicated as CG clusters, sharing concordant values of methDNA, Betadiff, and Corr, to generate
Integrated CG clusters (Figure 2C).
The scalable and open-source EpiMethEx 2.0 tool can be used for multilevel analyses of different methDNA and gene expression datasets, not only in cancer research but also for investigations in physiological and other pathological conditions. Moreover, EpiMethEx 2.0 package can be employed for comparisons between two or more groups, as well as for sample-by-sample analysis within the same group.
The Figure 2 was adapted from
EpiMethEx: a tool for large-scale integrated analysis in methylation hotspots linked to genetic regulation.